- | Academic & Student Programs Academic & Student Programs
- | Medical Innovation Toolkit Medical Innovation Toolkit
- | Regulation Regulation
- | Data Visualizations Data Visualizations
- |
How Productive Is the FDA’s Devices Program?
The purpose of Food and Drug Administration’s Devices and Radiological Health Program is to provide assurance of the safety, effectiveness, and quality of medical devices. The work of the Devices Program is carried out by the FDA’s Center for Devices and Radiological Health, plus field work done by the FDA’s Office of Regulatory Affairs (ORA). Most observers, and particularly the FDA, believe that user fees have been successful in helping the Devices Program meet its performance goals in reducing “the total time it takes to make decisions.” But the evidence presented here suggests that greater capacity and spending for the Devices Program over the last decade has not yet led to an increase in the number of new-product applications and reviews.
The purpose of Food and Drug Administration’s Devices and Radiological Health Program is to provide assurance of the safety, effectiveness, and quality of medical devices. The work of the Devices Program is carried out by the FDA’s Center for Devices and Radiological Health, plus field work done by the FDA’s Office of Regulatory Affairs (ORA).
Most observers, and particularly the FDA, believe that user fees have been successful in helping the Devices Program meet its performance goals in reducing “the total time it takes to make decisions.” But the evidence presented here suggests that greater capacity and spending for the Devices Program over the last decade has not yet led to an increase in the number of new-product applications and reviews.
Premarket approval applications (PMAs) for medical devices are for devices that are novel, meaning there is nothing on the market like them. The FDA is charged with making sure there is a “reasonable” assurance of safety and efficacy for these high-risk devices. For example, the artificial pacemaker is a medical device, as is a brain implant that allows a paralysis victim to move an artificial arm with her brain. These are clearly innovations that we want to encourage.
A goal of reducing decision-making time is not sufficient to increase the rate of innovations that cure or ameliorate health conditions. It could be the case, for instance, that fewer medical device inventions are being submitted to the FDA even though they are getting approved faster. The number of devices submitted for approval will be affected not only by the time taken to get a decision but also by the cost of the submission and the predictability of the decision.
The chart below plots the number of first-of-a-kind application submissions annually over the last decade, with inflation-adjusted spending on the FDA’s Devices Program. The accompanying table provides data on these and three additional measures of the Devices Program’s workload.
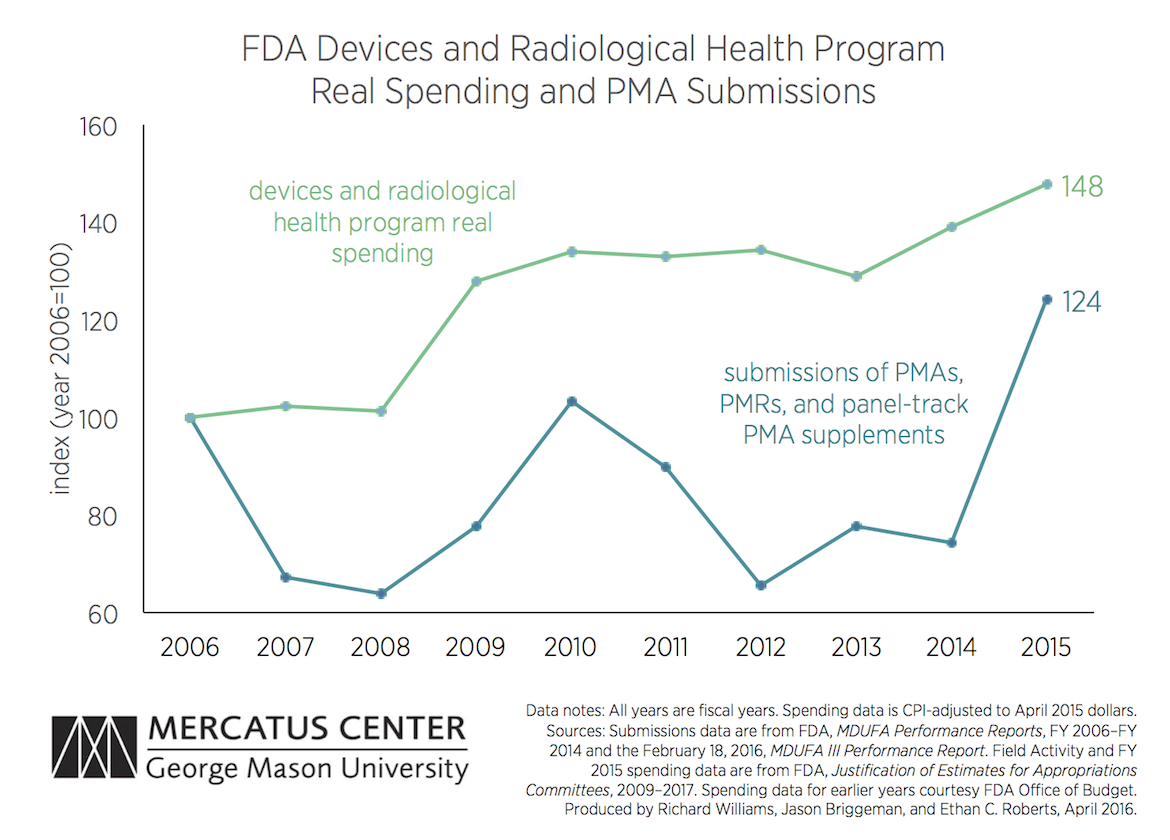
Annual real spending on the FDA’s Devices Program increased substantially in 2009 and has increased again since 2013, and it is now nearly 50 percent higher than in 2006. But while the number of first-of-a-kind device applications spiked last year (to 72, from 43 in 2014), in only two of the last nine years has the number of first-of-a-kind device applications been higher than it was in 2006.
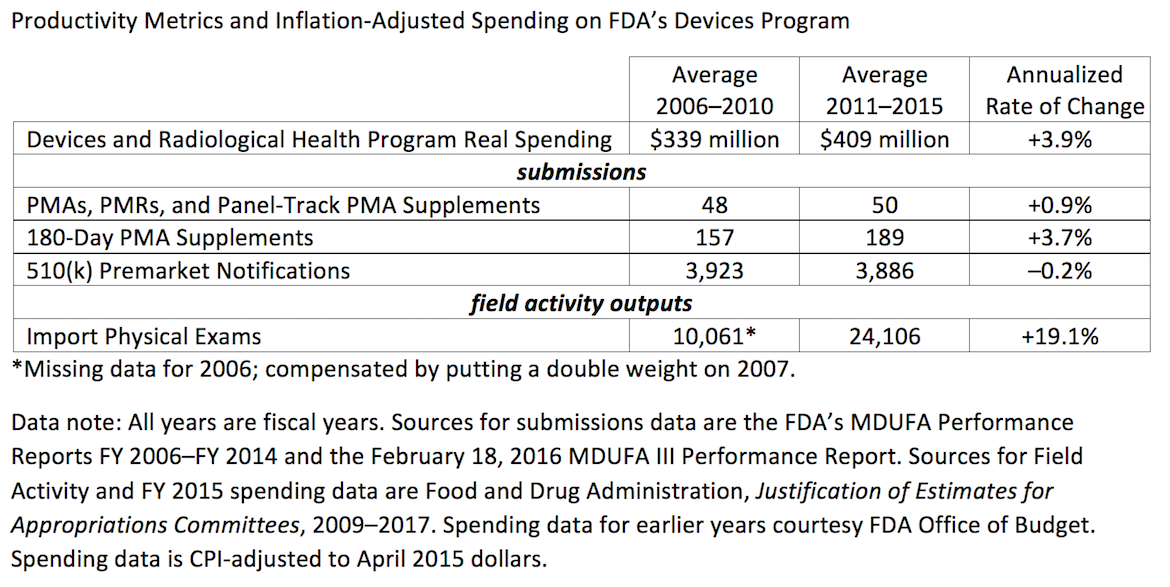
So the FDA was using substantially more resources to handle roughly the same number of device PMAs (although the number of PMA supplemental submissions was somewhat higher in the last five years than in the preceding five years). Resources may have been going to other activities, including increased examinations of imported goods (conducted by ORA) and incoming Medical Device Reports, which are user submissions regarding the safety of devices already on the market.
Despite more resources and the FDA’s Medical Device Innovation Initiative, it seems they have not yet “accelerate[d] innovation focused on high priority unmet health needs.”
To speak with a scholar or learn more on this topic, visit our contact page.

