- | Healthcare Healthcare
- | Policy Briefs Policy Briefs
- |
Regulators Could Allow Early Use of COVID-19 Vaccine or Treatment
Quickly developed therapeutic, prophylactic, and vaccine treatments will be essential to defeat the COVID-19 pandemic. Standard regulatory and development protocols typically yield drug development in 6 to 11 years. However, experimental vaccines and prophylactics against COVID-19 are in development and currently in trials, making deployment after safety trials feasible within months. Though efficacy of these treatments is uncertain, with probability of success, on average, at just 16 percent, efficacy can still be tested in real-world settings by adopting an approach that allows patients to begin drug treatments earlier in the regulatory process.
After phase I trials have been completed and data have been gathered, an expert committee should be convened to advise on the risk, benefit, and chance of success of the novel agent with a view toward distribution to the public through patient choice. There is limited downside risk and a good chance of a positive outcome with this approach. The US Food and Drug Administration (FDA) has the authority to approve pharmaceuticals requiring further trials, but the success of this approach will depend on coordination between sponsoring research organizations, trial designers, and regulators. International pharmaceutical regulators, such as those in the United Kingdom, Europe, or Japan, can also adopt this approach.
At this point, a clear indication of regulators’ openness to this approach would provide certainty for organisations and accelerate development of possible vaccines and treatments. When adjusting for probability, this approach may save hundreds of thousands of lives in the United States and more globally.
Background
On average, it takes between 6 and 11 years, from phase I to approval, to develop a new drug (see figure 1).

Part of what makes this process so lengthy is the regulatory burden of proving safety and then efficacy to the standard of random controlled trials, preferably in a double-blind format.
Critics of this process include (separately) Peter Thiel and Bartley Madden, among others. Madden has suggested that the regulatory pathway can be eased by allowing access to pharmaceuticals for patients earlier in the process. Madden calls this a dual-tracking process, with a patient free-to-choose track at phase II. The idea extends concepts present in the Right to Try Act (2018), though the act only applies to patients who have a life-threatening illness. This brief builds on these ideas specifically to deal with the challenges of COVID-19.
Current Experimental Treatments, Prophylaxis, and Vaccines
There are also several proposed therapeutics currently in trial for COVID-19. These agents are for (1) drugs currently approved for other indications and targeted at COVID-19 directly (e.g., chloroquine, baricitinib, ruxolitinib), (2) drugs currently not approved for any indication (e.g., remdesivir), and (3) drugs targeted at fatal complications of COVID-19 (e.g., tocilizumab). There are also therapeutics derived from plasma sources. The World Health Organisation (WHO) has detailed over 20 therapies in the first category.
There are several experimental vaccines in development, of which at least one—mRNA-1273—was, as of March 16, 2020, already in phase I safety trials. This phase I study will provide important data on the safety and immunogenicity of mRNA-1273. Immunogenicity means the ability of the vaccine to induce an immune response in participants. The open-label trial is expected to enroll 45 healthy adult volunteers ages 18 to 55 over approximately six weeks. There are at least two experimental prophylactic therapeutic regimes in process, one based on developing manufactured antibodies and one using plasma-derived antibodies from patients’ blood.
Proposals
There are two sets of proposals being considered, one for approved drugs in non-COVID-19 indications and another for unapproved treatments and vaccines. Trials should start now for already-approved drugs. If the originating companies are reluctant, either because the drugs have limited commercial value (e.g., chloroquine is generic) or because the commercial value of the drug is uncertain, (e.g., baricitinib, ruxulitnib) then the government should pay for the trials and forward-buy the drug on trial success. This policy of “advanced market commitment” has been successful in incentivizing research and development for combatting diseases common in low-income countries. According to the National Institutes of Health’s clinicaltrials.gov database, as of early March, only two therapeutic trials are running in the United States, remdesivir and mRNA-1273, and none are running in the United Kingdom. While product is available for most of the drugs on the list that the WHO has detailed as possible treatments, trials of these drugs are lacking. Regulators should allow compassionate use.
While treatment is important, prophylaxis through vaccines is arguably even more important. In the US, the FDA is responsible for vaccine development regulation enforcement. The FDA typically requires a biopharmaceutical company to complete the following steps in the development of a new vaccine:
- Investigational New Drug application
- Pre-licensure vaccine clinical trials
- Biologics License Application (BLA)
- Inspection of the manufacturing facility
- Presentation of findings to the FDA’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) external
- Usability testing of product labeling
For a US biopharmaceutical company, there are typically six stages in vaccine development:
- Exploratory stage
- Pre-clinical stage
- Clinical development
- Regulatory review and approval
- Manufacturing
- Quality control
The clinical development stage is a three-phase process. During phase I, small groups of people receive the trial vaccine as a test for safety. In phase II, the clinical study is expanded and the vaccine is given to people who have characteristics such as age and physical health similar to those for whom the new vaccine is intended. In phase III, the vaccine is given to thousands of people and tested for efficacy and safety. Many vaccines continue to phase IV, consisting of formal, ongoing studies after the vaccine has been approved and licensed.
A typical vaccine, when it has passed phase I, has a 16 percent chance of making it to the commercial market (see table 1). The vaccine still has risk from further safety, efficacy and regulatory hurdles.
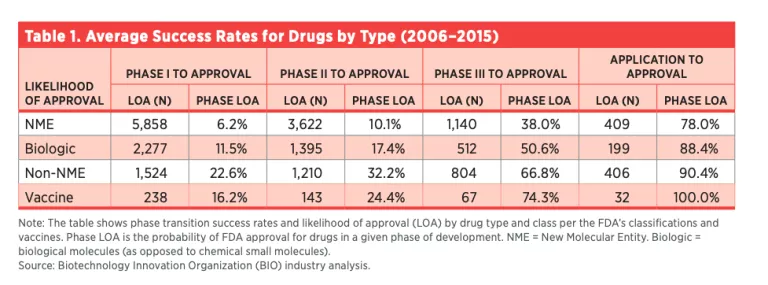
Even if the assessment of the vaccine at this stage is only the typical 16 percent chance of success, if safety downsides are considered limited, earlier dissemination of the vaccine would equate to a potential probability-adjusted 220,000 US lives saved, if the projected scenario of 1.4 million deaths proves accurate (17 percent of the older-than-65 US population = 55 million, 50 percent infection rate = 27 million, and 5 percent crude death rate = 1.375 million).
Manufacturing of the vaccine would have to scale up considerably in Q2 2020, but with investment and dedication, it should be possible for commercial organizations to succeed.
A similar strategy can be developed for different vaccine programs running in parallel. This idea could also extend to the development of prophylactic antibody treatments, which are expected to enter phase I trials in Q3 2020. These phase I trials could produce data by early 2021, and a similar expert committee could assess the data then. A typical biological success rate is 11 percent after phase I, which would still result in a probability-adjusted hundred of thousands of lives saved. This treatment may be more expensive but still within a $10 billion budget.
This brief focuses on the United States, as the United States has a large population and the resources to enact these proposals. Similar ideas, however, could be enacted in Europe, Japan, the United Kingdom, and other jurisdictions that have similar regulatory frameworks.
Risks and Tradeoffs
There may be further safety risks that a phase I trial would not assess. Multiple phase I trials would improve the statistical insights into the data. These insights can be discussed by the expert committee, and individuals can make their own choice based on the best available data. It may turn out that the vaccine is ineffective, which may alter patient behaviors. These risk assessments can be determined by the appropriate scientists and policymakers. A moral philosopher or ethics expert as well as representatives of the general public may be allowed on the committee. There are precedents for using a Compassionate Use Advisory Committee model.
All pharmaceutical agents have benefit and risk. This author notes that pharmaceutical regulators have been under intense pressure from antivaccination campaigners. Furthermore, drug regulators suffer from a principal-agent challenge. In ordinary times, regulators take much of the blame for safety problems but receive little of the credit for positive lifesaving outcomes. As a result, regulators (the agents) may become more risk-averse than the informed general public (the principals).
One argument in favor of regulation is that knowing whether drugs are safe and effective is something far beyond the common knowledge of patients. For typical diseases in ordinary times, this reasoning might hold much weight. However, in the face of an extraordinary medical need and a pressing timeline, the balance of risk and reward is different. Therefore, an exception should be made for a potential COVID-19 vaccine or prophylactic.
Money will need to be spent. A rough, back-of-the-envelope estimate of $1 billion to $2 billion per vaccine candidate is not unreasonable, but there will be innovation knowledge and manufacturing assets built with this investment, regardless of success. Given the likely scale of the economic damage, ranging in the hundreds of billions to the trillions of US dollars (based on simply cutting 2 percent of global GDP or more), the investment tradeoff is favorable.
Bold Measures for Difficult Times
Government should, in collaboration with pharmaceutical companies and researchers, consider accelerated regulation and trial design for novel agents against COVID-19. The data for any vaccine or therapeutic should be assessed after phase I, and based on that assessment, it should be considered a patient choice to take the experimental agent. At 16 percent probability of efficacy, after phase I, a vaccine would save, adjusting for probability, potentially hundreds of thousands of lives with limited downside risks.
About the Author
Benjamin Yeoh is a healthcare investor with 18 years of experience. In addition to managing pension fund assets, Yeoh sits on advisery committees to the UK financial regulator, the Financial Reporting Council (FRC), and the International Accounting Standards Board (IASB). He is an associate fellow of the Hoffman Centre at Chatham House. He is not a virologist or an infectious disease expert, and this brief was written in his personal capacity. The views in this brief are expressed in good faith and should be investigated by those with expert knowledge. These ideas are suggested for public health reasons only. No endorsement of any company mentioned for investment purposes is made or implied. Yeoh’s blog is at thendobetter.com

